2001 Summer School on Computational Materials Science
Tools for multiple length and time scales
![]() Last updated July 18, 2001 We introduced the tools and concepts used in first principles simulations of atomic and molecular systems: the use of the fundamental laws of matter with high performance computing to enable predictions of experimental properties of materials. The basic simulation and electronic structure methods were covered in the first week. In the second week, recently developed methods for handling systems with longer time and of larger size were discussed.
Last updated July 18, 2001 We introduced the tools and concepts used in first principles simulations of atomic and molecular systems: the use of the fundamental laws of matter with high performance computing to enable predictions of experimental properties of materials. The basic simulation and electronic structure methods were covered in the first week. In the second week, recently developed methods for handling systems with longer time and of larger size were discussed.
Topics and presenters
| Presenter name | Link to the presentation file |
|---|---|
| Noam Bernstein Naval Research Laboratory | Coupling Molecular Dynamics and Continuum Methods |
| Tim Germann Los Alamos National Laboratory | Accelerated Molecular Dynamics Methods |
| Jan Jensen University of Iowa | Quantum Mechanics / Molecular Mechanics Methods |
| Jeongnim Kim Ohio State | Object-oriented programming |
| Blair Tuttle Pennsylvania State University, Behrend | Point Defects in Semiconductors |
| David Ceperley University of Illinois | Introduction to Computer Simulation of Atomic-Scale Systems |
| Eric de Sturler University of Illinois | Iterative Methods for Linear, Non-linear, and Eigenvalue Problems |
| Duane Johnson University of Illinois | Kinetic Monte Carlo: Bare Bones and a Little Flesh |
| Richard Martin University of Illinois | Density Functional Theory |
| Todd Martinez University of Illinois | Basics of Quantum Chemistry |
Outlines of Presentations
Noam Bernstein
Title: Coupling molecular dynamics and continuum methods
- Overview of MD and continuum mechanics
- Concepts, variables
- Strains, stresses
- Displacements
- Finite elements, maybe boundary elements
- Simple expression for E = 1/2 (U K U + U^. M U^.)
- Compare concepts
- Atomic positions vs. fields
- Positions -> displacement field
- Asides:
- Fluids: velocity fields
- Approaches
- Gumbsch, Hoover, CLS
- Mesh refining
- Slave particles
- Slave particles/displacements
- Coarse grained MD
- Quasi-continuum
- Outstanding issues
- Variable constitutive laws (CGMD)
- Temperature/vibrations
- Fluids (incompatible fundamental variables)
David Ceperley
Title: Introduction to Computer Simulation of Atomic-Scale Systems
Background Article:
"Microscopic simulations in physics", D. M. Ceperley, Rev. Mod. Phys. 71
S438 (1999).
- Introduction
- statistical mechanics,
- simulation methods,
- ergodicity,
- statistical errors
- Molecular dynamics
- integrators,
- boundary conditions,
- long range potentials,
- thermostats,
- order parameters,
- static and dynamic properties
- Monte Carlo methods:
- Simple Monte Carlo
- Markov chains,
- transition rules
- Brownian Monte Carlo
Eric de Sturler
- Krylov subspace methods for solving large sparse linear systems of equations
- intro iterative methods
- GMRES, MINRES, CG
- Convergence bounds and how to use them
- BiCG and related methods
- Preconditioning
- Krylov subspace methods for large sparse eigenvalue problems
- Arnoldi / Lanczos
- Davidson / Jacobi-Davidson
- Multigrid methods
- basic iterative methods and smoothing
- smoothing analysis for Fourier modes
- grid transfer operators and multigrid
- local mode analysis
- Fast Multipole Methods
Tim Germann
Title: Accelerated Molecular Dynamics Methods Background Article:
"A method for accelerating the molecular dynamics simulation of infrequent events", A. F. Voter, J. Chem. Phys. 106 4665 (1997).- Background (15 min)
- Rare event examples: epitaxial growth, radiation damage - e.g. plutonium aging due to self-irradiation, ...
- Pitfalls of KMC: concerted multiatom mechanisms, unconventional crystal structures or grain boundary diffusion, ...
- One possibility: Henkelman-Jonsson saddle-point enumeration + KMC Seek MD acceleration technique(s) w/o biasing the dynamics
- Hyperdynamics (75 min)
- Basic idea: boost PES in wells w/o affecting saddle points Mathematical justification (assuming TST) & definition of an accelerated time
- Demonstration on model system(s) using simple boost functions
- More sophisticated boost functions
- Application to more complex systems
- Unsolved problems: extension to systems with low barriers (or a range of barriers, e.g. glasses, liquids, proteins)
- Related approaches: temperature accelerated dynamics (15 min)
- Basic idea: increase simulation temperature to find neighboring saddles, but reject transitions until low-temperature path can be accepted
- Mathematical justification (assuming harmonic TST)
- Demonstration on model system(s)
- Related approaches: parallel replica dynamics (15 min)
- Basic idea: independent realization of system on each processor to explore phase space more rapidly; first processor to find escape pathway is accepted
- Mathematical justification (assuming infrequent events)
- Demonstration on model system(s)
- Combining II and IV -> parallel replica hyperdynamics (15 min)
- Multiplicative boost achieved
- Example: epitaxial growth of Cu/Cu(100) on 1000 procs -> 0.3 s
- Outlook & Prospects (15 min)
- Lab
will focus on *'d topics; vary boost function, system size and complexity (e.g. single adatom diffusion vs. islands with both low and high barriers) to demonstrate where hyperMD works well and where it runs into difficulties.
Jan Jensen
Title: QM/MM MethodsBackground Articles:
"The Effective Fragment Potential Method: A QM-based Approach to Modeling Environmental Effects in Chemistry", M. S. Gordon, M. A. Freitag, P. Bandyopadhyay, J. H. Jensen, V. Kairys, and W. J. Stevens, J. Phys. Chem. A 105 293-307 (2001).
"A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments", R. B. Murphy, D. M. Philipp, R. A. Friesner, J. Comp. Chem. 21 1442-1457 (2000).
"Hybrid potentials for large molecular systems", P. Amara and M. J. Field, Computational Molecular Biology Series title: Theoretical Computational Chemistry (J. Leszczynski, Ed) vol 8. pg 1-33 (1999) (Amsterdam; New York: Elsevier).
- Overview and History
- Origin/Philosophy
- Main Approaches
- Commercial/Public Codes
- Methodological Considerations
- QM Description (semiempirical vs ab initio)
- MM Description (charges vs. multipoles, polarization, short-range interactions)
- Covalent Boundary
- Mechanical vs. Electrostatic Coupling
- Vibrational Effects/Dynamical Issues
- Bulk Solvation
- Applications
- Main Areas
- Specific case studies: Biochemistry
- Specific case studies: Material Science
- Outlook/Future Directions
Duane Johnson
Title: Kinetic Monte Carlo: Bare Bones and a Little FleshBackground Article: "Theoretical foundations of dynamical Monter Carlo simulations", K.A. Fichthorn, J. Chem. Phys. 95 1090 (1991).
- Introduction to Kinetic (a.k.a. Dynamical) MC (5 min.)
- Time Scales: MD, MC, and KMC
- A Hybrid Monte Carlo: MD + KMC
- Solution of the "Markovian" Master Equation (15 min.)
- MC vs. KMC
- Detailed Balance Requirements
- Rudiments of KMC (simulation time vs. real time) (45 min.)
- Dynamical Hierarchies (time scales via transition rates)
- Poisson Processes (an necessary aside) - examples: radiation counts, binomial distributions
- Simple Example: Adsorption-Desorption in 2-D (see background article)
- Generalization
- Algorithms for "Fast" Monte Carlo (30 min.)
- Basics of the "n-fold-way" (or "residence-time") algorithm B. Improvements using Binary or K-search Methods
- Laboratory KMC Exercise (20 min.)
- Vacancy-assisted Ordering in 2-D Binary Alloy
- Special Effects: radiation damage via ballistic jumps
- Beware of Solving Master Equation via Approximations (if time permits, 10 min.)
- Technical Point: Fokker-Planck Equation is wrong.
- Technical Point: First-order Fix-up from Kubo
- Example: Non-equilibrium transitions in fcc AB3 compounds, see Haider, Bellon and Martin, Phys. Rev. B 42 8274 (1990)
Jeongnim Kim
Title: Object-oriented programming- Introduction:
- motivation for modular programming
- basics of Object-oriented programming
- requirements: Hamiltonian, spatial layout, algorithms
- communicating software design: class diagrams, documentations
- Practical implementation of OO programming in MD simulations
- Design Patterns
- Templates
- Parallel computing and OO framework
Richard Martin
Background Articles:
"Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients", M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias, and J. D. Joannopoulos, Rev. Mod. Phys. 64, 1045-1097 (1992).
"Pseudopotential Methods in Condensed Matter Applications", W. E. Pickett, Computer Physics Reports 9, 115 (1989).
More specialized article: "Linear Scaling Electronic Structure Methods ", S. Goedecker, Rev. Mod. Phys. 71, 1085-1123 (1999).
- Basic Density Functional Theory & Bands in Crystals
- Hohenberg-Kohn theorems
- Meaning for the functional - Levy-Lieb variational principle
- Practical DFT: the KOhn-Sham approach
- Solving the self-consistent Kohn-Sham equations
- Examples of results for simple systemsm - successes, failures
- Pseudopotentials and efficient iterative methods for calculations
- Calculations for atoms; generation of ab intio pseudopotentials Semilocal and Kleinman-Bylander non-local separable operators
- Bloch theorem and bands for electrons in periodic crystals Definition of the crystal stucture and Brillouin zone in programs the will be used in the lab
- Plane wave DFT codes Empirical pseudopotentials: examples of band structures Ab intio pseudopotential calculations Achieving self-consistency Examples of ab intio results
- Iterative methods General aspects: Krylov subspaces; iterative solutions Solution by minmization: Conjugate gradradient methods Must impose constraint of orthonormality Solution by residual minimization (Connection to VASP code that will be used by Tuttle)
- Car-Parrinello quantum molecular dynamics
- Order-N methods
- Locality and quantum mechanics The density matrix Wannier functions (generalized)
- Methods for solving equations taking advantage of locality A new functional that can be minimized without the constraint of orthonormality Density matrix methods - "McWeeny purification" Fermi operator projection methods Combinations of the methods
- Examples of results Giant Fullerenes Simulation of fullerenes crahing into diamond surfaces Hot metals A complete turn of DNA
- Lab: Working with simple codes for atoms, empirical pseudopotential and tight-binding. Application to Si, GaAs.
- Structure of modular F90 code
- Physical problems: Changes in the bands with volume Phonon displacenents
Todd Martinez
- Fundamentals of Potential Energy Surfaces
- Stationary Points, Minima, Transition States, Intersections
- Analytic Derivative Methods
- Basic Optimization Techniques
- Transition State Theory
- Equilibrium Formulation
- Phase Space / Dynamical Formulations
- Quantum Mechanical Variants
- Dynamic Corrections / Tunneling Corrections
- Multiple Time-Scale Integrators for Classical Dynamics
Blair Tuttle
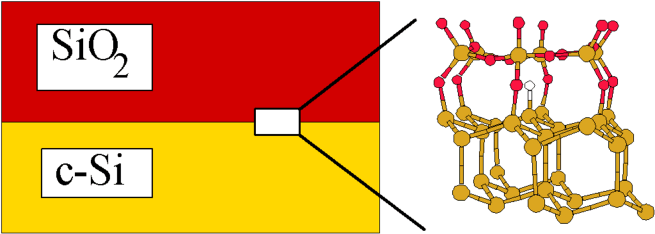
Title: Point Defects in Semiconductors: Focus on H in Si, SiO_2 and at their junctions- Supercell vs Cluster calculations
- Basis set convergence
- Cell size convergence
- K-point sampling
- Structural properties
- H clusters in a-Si
- Hyperfine calculations
- Si_db at Si-SiO_2 interface
- Atomic H in c-Si
- Electrical Activity
- Examples
- Si_db in a-Si
- Atomic H in c-Si and SiO_2
- (band gap problem and some solutions)
- Dynamical Properties
- Vibrational spectra
- Atomic H in Si and SiO_2
- (IR spectra)
- Diffusion
- Atomic H in Si and SiO_2
- (elastic nudge method)
- Overview
Lab: use atomic H in c-Si for most of the calculations.
Photos









Sponsors
The Summer School was held at the Beckman Institute on the University of Illinois, Urbana-Champaign campus and was sponsored in part by:
- Materials Computational Center
- National Center for Supercomputing Applications
- Department of Physics
- Frederick Seitz Materials Research Laboratory
- Computational Science and Engineering Program
- NSF Combined Research-Curriculum Development Program